Abstract
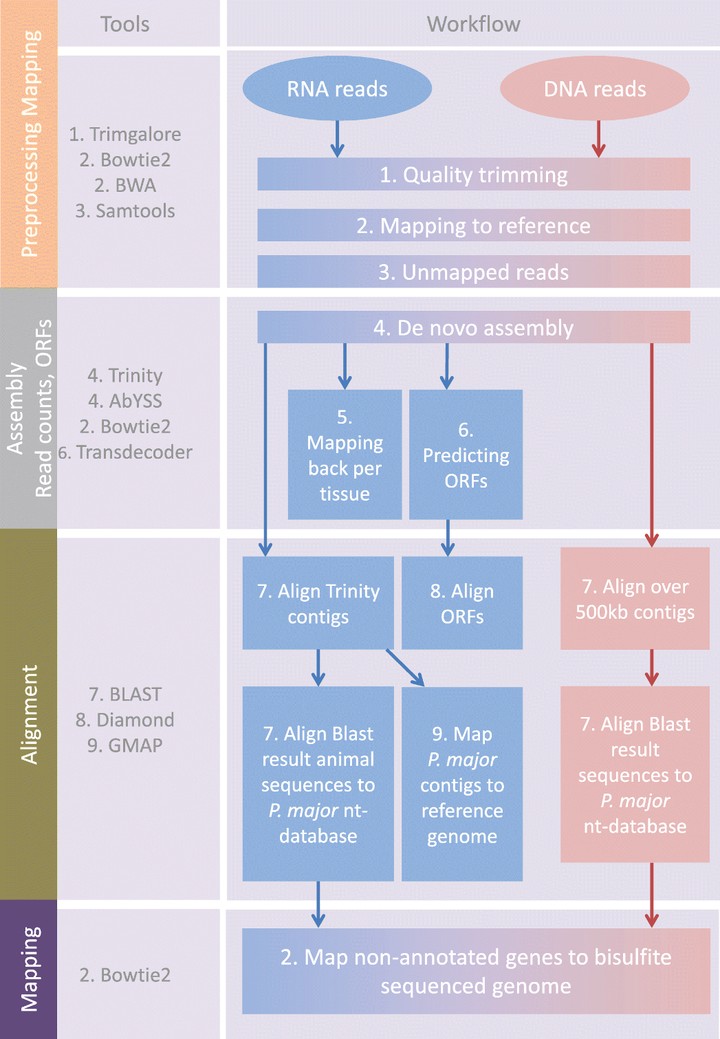
A widely used approach in next-generation sequencing projects is the alignment of reads to a reference genome. Despite methodological and hardware improvements which have enhanced the efficiency and accuracy of alignments, a significant percentage of reads frequently remain unmapped. Usually, unmapped reads are discarded from the analysis process, but significant biological information and insights can be uncovered from these data. We explored the unmapped DNA (normal and bisulfite treated) and RNA sequence reads of the great tit (Parus major) reference genome individual. From the unmapped reads we generated de novo assemblies, after which the generated sequence contigs were aligned to the NCBI non-redundant nucleotide database using BLAST, identifying the closest known matching sequence. Many of the aligned contigs showed sequence similarity to different bird species and genes that were absent in the great tit reference assembly. Furthermore, there were also contigs that represented known P. major pathogenic species. Most interesting were several species of blood parasites such as Plasmodium and Trypanosoma. Our analyses revealed that meaningful biological information can be found when further exploring unmapped reads. For instance, it is possible to discover sequences that are either absent or misassembled in the reference genome, and sequences that indicate infection or sample contamination. In this study we also propose strategies to aid the capture and interpretation of this information from unmapped reads.
Veronika N. Laine
Post doc
My research interests include molecular biology and evolutionary and behaviour genetics.